The importance of reaction modeling in new drug development
Bringing new drug products to market is a continuously increasingly expensive and time-consuming process which is compounded by the need to bring products to market faster. Therefore, a plethora of tools and strategies, such as automation, running development stages in parallel and reaction modeling, are used to fast-track drug molecules at R&D stage and provide a kickstart to later stage development. Reaction modeling is able to provide in-depth understanding of the reaction mechanisms, kinetic understanding about important factors for optimization, as well as how the reaction will respond during scale up.
To construct these models, you need high quality data at scales relevant to the process, which in the past has limited when they could be developed (as data rich experimentation is not possible on high throughput experimentation).
However, ReactALL provides an automated platform for generating medium-throughput, data-rich experimentation with little material, allowing for earlier and faster development. This is demonstrated in the work by Argüelles et al [i], where a new workflow was designed and tested to develop a model from as few as 6 experiments. ReactALL combines the throughput of high-throughput (HT) systems with the data richness of low-throughput (LT) approaches, delivering a powerful medium-throughput (MT) solution.

The new modeling approach
The model was developed for an acid catalyzed acetal deprotection of molecule 1 using acetonitrile/water mixture as solvent and p-toluenesulfonic acid monohydrate (TsOH) as the catalyst, summarized in Figure 1. Using this ReactALL a baseline experiment was conducted, followed by 4 experiments varying temperature, starting concentrations and TsOH volume, and a model validation. Using the automated sampling capabilities of the ReactALL to collect 8 samples from each experiment then analysis by HPLC to track the reaction results summarized in Figure 2. The results were then processed with Reaction Labs modeling software.
Figure 1. Acetal Deprotection of Compound 1 [Org. Process Res. Dev. 2026, 30, 2, 363–371. Copyright © 2026 American Chemical Society]
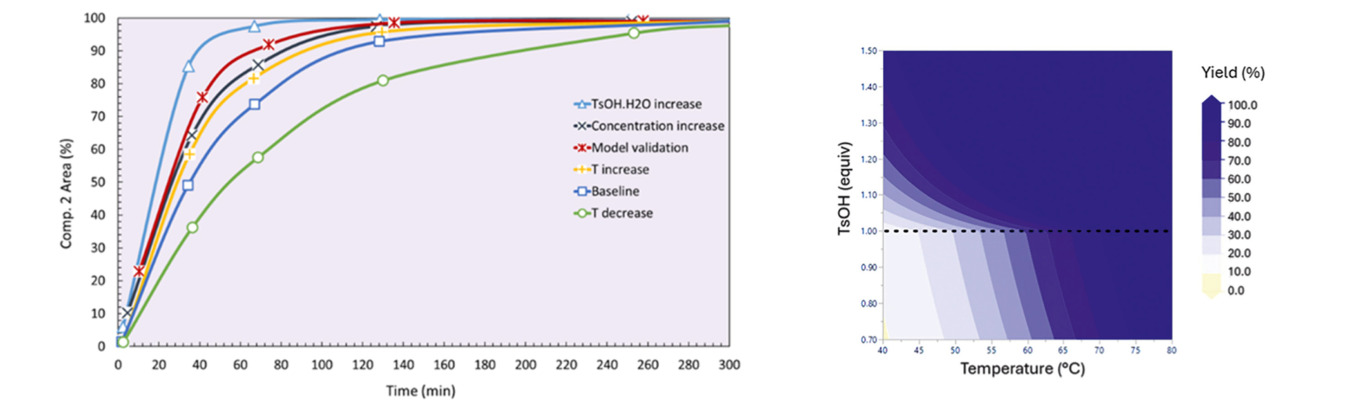
Figure 2. Results of screening reactions (Left) and surface response plot from model (right) [Org. Process Res. Dev. 2026, 30, 2, 363–371. Copyright © 2026 American Chemical Society]
Three mechanistic models were tested, and a surprising result was found that the quinolinium functional group was involved in catalysing the reaction. From this model a surface response plot was created to show operating zone where the yield of the reaction could be optimized. Additionally, a scaleup experiment was conducted at 20X scale factors and was found to match the prediction from the model. The overwhelming success of this workflow has led to it being implemented in other ongoing product reactions.
Conclusion
Modeling a chemical reaction is a powerful way to speed up development of a new molecule, providing mechanistic, kinetic and scale up information. However, they require data rich experimentation that is not possible on many platforms. A new workflow was presented utilizing the ReactALL medium throughput and the automated sampling to screen conditions and variables to build and validate a kinetic model from only a few experiments. The model revealed the surprising role of the quinolinium species in the catalysis. Additional plots were generated that allow for reaction optimization and scaleup. This new workflow has since been applied to other reaction scheme demonstrating the level of reaction and process understanding enabled by ReactALL.
Reference
[i] Expedited Kinetic Modeling Workflow Leveraging Medium-Throughput Automation, Stefanie Swiecki and Alonso J. Argüelles, Organic Process Research & Development 2026 30 (2), 363-371, DOI: 10.1021/acs.oprd.5c00373
Curious to learn more?
Download the full application note "Robust and scalable kinetic model generation from automated small scale experimentation".